For the final project in my Spring 2009 Perspectives in Biological Engineering class at MIT, I implemented a computational model of human nucleotide excision repair, then wrote this research proposal based on the results of that model. Download a PDF copy here.
A. Specific Aims
Nucleotide excision repair (NER) is a complex pathway that is responsible for the repair of bulky adducts in bacterial and eukaryotic DNA [1,5]. Defects in NER are responsible for a variety of human diseases, and upregulation of NER has been implicated in resistance to treatment in cancers [2,7]. Despite a wealth of structural and biochemical information about the components of the pathway, our understanding of the molecular mechanism remains limited [8]. Developing a mechanistic view of NER would provide valuable insight into a variety of human diseases.
Specific Aim 1: Construct and validate an improved mathematical model of human NER that incorporates mechanistic kinetic proofreading
An ordinary-differential-equation-based model of human NER was recently published that accounts for the high specificity of NER through kinetic proofreading [3], a common feature in biological systems that provides greater specificity between two substrates than would be predicted based purely on thermodynamics [4]. This model accounts for kinetic proofreading in a phenomenological manner that relies on non-biological free parameters. We will improve upon this model by introducing mechanistic kinetic proofreading that is based on experimentally testable parameters, and we will validate our model by comparison to the same in vitro NER assays as the original.
Specific Aim 2: Obtain biologically-based model parameters through experimental manipulation
A variety of in vitro analytical techniques exist for the determination of kinetic and thermodynamic parameters such as K_d, k_on, and k_off. Using isothermal calorimetry, surface plasmon resonance, and/or fluorescence-based assays, we will determine physiological values for kinetic proofreading parameters in human NER.
Specific Aim 3: Validate model hypotheses in an in vitro cell-free model
Our model will result in specific predictions about the affinities and stabilities of various interactions and complexes within the NER pathway. A variety of well-characterized cell-free assays exist for testing such predictions in vitro. We will use standard chemical biology techniques (site-directed mutagenesis, kinase inactivation, fluorescent probes) to validate the hypotheses generated by our improved model.
Specific Aim 4: Validate in vitro predictions in an in vivo whole-cell system
The molecular-level hypotheses that we examine in an in vitro model will translate to predictions of observable phenotypic changes in an in vivo system. That is, the results of Specific Aim 3 will generate predictions about the genomic stability and sensitivity to (photo)chemical damage of a whole-cell system. Using an establlished eukaryotic model with standard survival and DNA damage assays, we will validate the predictions of our in vitro studies.
B. Background & Significance
Every day, our DNA is subjected to a variety of endogenous and environmental stresses, including reactive oxygen species, ultraviolet (UV) light, and industrial chemicals. Many of these stresses cause damage to DNA that can result in mutations and, ultimately, genomic instability and disease [28,29]. The human body has developed a variety of ways to protect DNA from damage and mutations, including direct reversal, base proofreading, and nucleotide excision repair (NER).
NER in particular is essential to human life for a variety of reasons. It is the only completely general purpose method of repair, meaning it is the only way to repair damage products resulting from novel chemicals (such as industrial toxins) [30]. It is also a major source of repair of oxidative damage, which is an unavoidable byproduct of an oxidative metabolism [31]. Finally, NER is the major source of repair of cyclobutane pyrimidine dimers and so-called 6-4 photoproducts, which are the main lesions induced in human cells exposed to UV light [5]. The structures of these damage products are shown here. Given these myriad functions, it is no surprise that defects in NER cause a variety of diseases, including xeroderma pigmentosum and Cockayne syndrome [2]. In addition, given its role in repairing damage from exogenous chemicals, it is understandable that upregulation of NER has been linked to resistance to chemotherapy and radiation in human cancers [7]. Studying NER and its defects therefore has a direct role in human health.
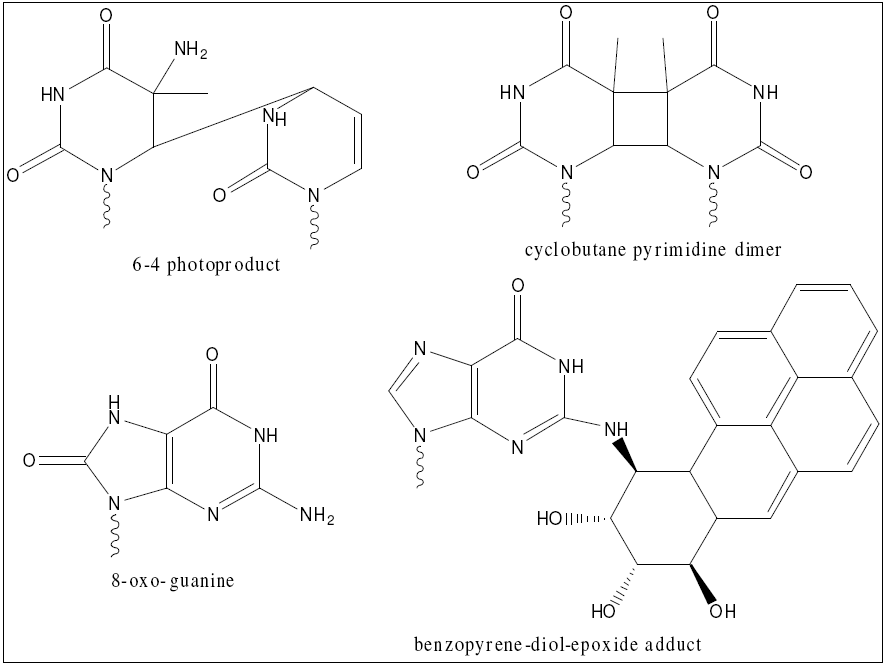{kind=link}
In humans, NER comprises over 30 proteins that function collectively to form the complete pathway [6]. However, the system can be functionally reconstituted with only six factors: RPA, XPA, XPC-TFIIH, XPG, and XPF. These work together to perform the steps in NER, which are well-defined and highly conserved. First, damage recognition proteins (RPA, XPA, and XPC-TFIIH) bind to the damaged DNA, forming a relatively unstable open complex. This binding step displays cooperativity, enhancing both the speed and specificity of assembly. ATP hydrolysis converts this into the stable preincision complex 1 (PIC1), and TFIIH (a multiprotein complex) locally unwinds 25-32-bp, roughly symmetrically distributed over the site of damage. XPG then displaces XPC in an ATP-dependent manner to form PIC2. In another ATP-dependent step, XPF is recruited to form PIC3. PIC3 rapidly proceeds to the excision step, where XPG cuts 2-8-bp 3' to the damage and XPF cuts 15-24-bp 5' to the damage. Following excision, all proteins except RPA dissociate, and the resulting single stranded gap is repaired by gap-filling synthesis [32].
An essential feature of NER is that it displays remarkable specificity, in that it rarely excises undamaged DNA. In fact, the error rate for NER is such that it cannot be accounted for simply by invoking the difference in affinity of the damage recognition proteins for damaged versus undamaged DNA; the corresponding free energy difference is insufficient to explain the low error rate. In general, a single "test" based on affinity should fail p fraction of the time, with
p = e - ΔF / kT
where ΔF is the difference in free energy between the two interactions and kT is a scaling factor (Boltzmann constant time temperature) [8]. However, as more "tests" are performed, the fraction of failures goes as pn, where n is the number of comparisons. This fact is exploited by the NER system in a process known as kinetic proofreading, which was first proposed to explain a similar problem in protein synthesis [4]. Kinetic proofreading is accomplished by introducing a number of irreversible steps between the initial association (proteins binding to DNA) and the final reaction (dual incision of DNA). Each irreversible step represents another "test" of the system, and collectively they serve to create a sort of molecular time delay that helps prevent accidental incision of undamaged DNA. The complete pathway is illustrated in this figure [33].
![illustrated in this figure [33]](images/ner_2.png){kind=link}
While the steps in NER are well-understood, the molecular mechanism of these steps and the exact functions of the proteins involved are not well characterized (as evidenced by the disparity in the number of proteins needed to reconstitute the system and the number known to be involved). In particular, details of the kinetic proofreading, substrate specificity, and repair speed of NER are not well understood. This is an area where mathematical modeling can be of great utility because of its ability to generate testable hypotheses based on proposed mechanisms.
A model was recently published by Kesseler et al. that incorporates all the major features of pathway illustrated above, and accurately recapitulates the results of in vitro repair assays [3]. The network schematic of their model [3] is illustrated. Despite its success, the model relied on a number of nonbiological free parameters, making it difficult to draw inferences with a high degree of accuracy. In particular, the kinetic proofreading - one of the least well understood parts of NER at a mechanistic level - is implemented in a purely phenomenological manner. The model could be improved by introducing a more mechanistic treatment of the kinetic proofreading, then using that model to inform testable hypotheses with the ultimate goal of elucidating the exact mechanism of proofreading. Such an understanding could have potential impacts on the treatment of a variety of human diseases, including cancer. An accurate model of human NER would allow for the rapid simulation of hundreds of potential perturbations to the system, which in turn would reveal "weak points"; that is, components of the system that are most easily disrupted. These weak spots could then be used as leads for the development of selective inhibitors of NER. A general precedent for this strategy is seen in the fact that cells expressing high levels of an HMG protein are deficient in NER repair of cisplatin adducts and display increased sensitivity to killing by cisplatin [7]. The ultimate downstream goal of this work is to develop compounds that could be delivered in combination with existing chemotherapies as adjuvants to improve the therapeutic response in highly resistant cancers, thus providing a clear link to human health and rationale for this study.
![The network schematic of their model [3]](images/ner_3.png){kind=link}
C. Preliminary Data
To verify that our modified model maintained the accuracy of the original, we ran a simulation using parameters identical to those used in an in vitro NER assay by Kesseler et al. That assay resulted in repair of 12.5% of the total DNA after 90 minutes, and as can be seen in the resulting graph, our improved model mimics this behavior. We therefore hypothesize that our mechanism of kinetic proofreading (shown in D. Experimental Design) accurately reflects the elementary molecular interactions in human NER. We further hypothesize that analytical techniques will allow us to determine the actual values of the parameters above, thereby enhancing our understanding of kinetic proofreading. This hypothesis will be addressed in Specific Aim 2.
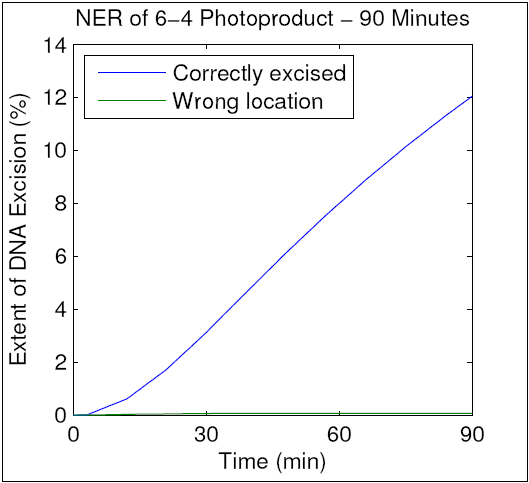{kind=link}
To better understand the topology of the network with regard to damaged DNA, we ran the simulation until repair plateaued at roughly 27% (similar to the original model) and tracked the concentration of the preincision complexes with damaged DNA bound. As can be seen in the resulting graph, PIC1 and PIC2 are formed and consumed within the first 60 minutes of repair, whereas PIC3 accumulates and converts to excised DNA much more slowly. We hypothesize that this reflects greater stability of PIC3 compared to PIC1 or PIC2, and that this stability may constitute a more stringent "final check" that has evolved to prevent accidental excision of undamaged DNA. Given that XPF is the only protein unique to PIC3, it is likely that XPF's affinity for damaged DNA mediates this increased stability. We hypothesize that XPF mutants with decreased affinity for DNA will display decreased PIC3 stability, dysfunctional NER, elevated levels of excision of undamaged DNA, and sensitivity to chemical- and UV-induced DNA damage. These hypotheses will be tested in Specific Aims 3 and 4.
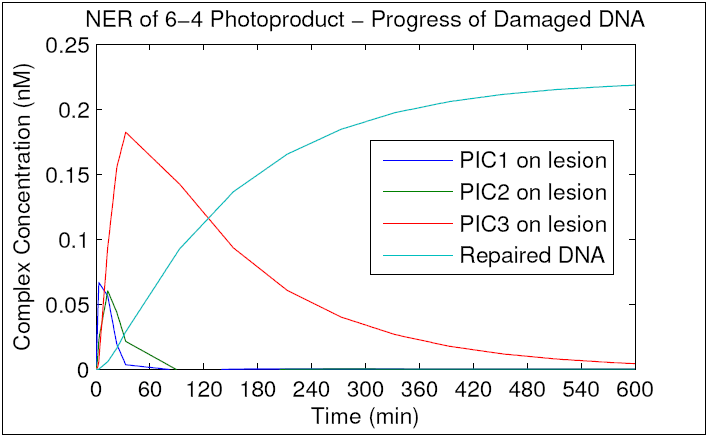{kind=link}
To further characterize the function of PIC3, we repeated the simulation and tracked the concentration of PIC3 for all three substrates. As can be seen in the resulting graph, almost no PIC3 forms away from the site of damage or on undamaged DNA. This is in contrast to PIC1, a large fraction of which is formed away from the site of damage. It is possible that he three PICs may have evolved their particular order and affinities to "specialize" in preventing different types of accidental excision based on the relative likelihood of the mistake (i.e. missing a lesion is more likely than binding to an undamaged strand). We hypothesize that PIC1 has relatively high stability when bound to undamaged DNA, compared to PIC2 and PIC3. Again, given that XPC is the only protein unique to PIC1, we hypothesize that XPC's affinity for undamaged DNA mediates this stability. We hypothesize that XPC mutants with decreased affinity for undamaged DNA will display decreased PIC1 stability, dysfunctional NER and sensitivity to DNA damage. These hypotheses will be tested in Specific Aims 3 and 4.
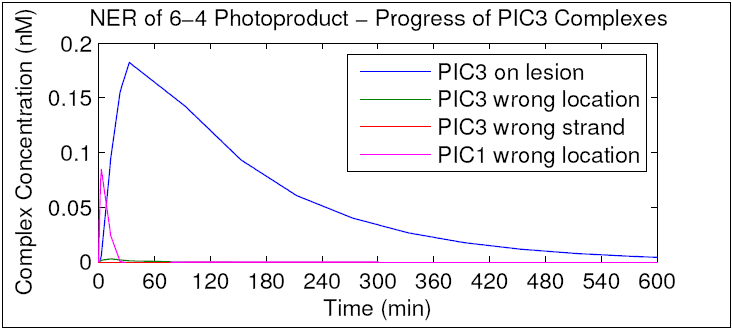{kind=link}
D. Experimental Design
D.1 Construct and validate an improved mathematical model of human NER that incorporates mechanistic kinetic proofreading
Kesseler et al. published a model that accurately recapitulates the NER activity seen in a cell-free assay using 6-4 photoproduct substrates [3]. This model took advantage of two concepts to explain the speed and specificity of NER: cooperative binding and kinetic proofreading. Cooperativity is a well established feature of a variety of biological systems, and its mechanistic details are well understood. Less commonly encountered is kinetic proofreading, a concept first proposed to explain the accuracy of protein synthesis, which has an error rate significantly smaller than that predicted by the difference in free energy between correct and incorrect tRNAs binding to the ribosome [4]. To account for this difference, a number of irreversible steps (most commonly protein phosphorylation of ATP hydrolysis) are inserted between the initial binding and the final effector step. This effectively creates a time delay because at each intermediate, the only options are to proceed or dissociate and start the process over entirely. This action can exponentially increase the "effective" difference in free energy between two species, thereby accounting for specificity.
While the general scheme of kinetic proofreading is well understood, its detailed mechanism is unique to a given system. In the case of NER, there are three kinetic proofreading steps that are dependent upon ATP hydrolysis. After each step there is a preincision complex (PIC) that can either fully dissociate or proceed to the next step. Kesseler et al. treated kinetic proofreading in a phenomenological manner by specifying that 10% of each of the reactions would lead to dissociation rather than the subsequent complex. While this model recapitulates experimental NER, it is not mechanistic and is not based on biologically founded or testable parameters.
To improve our understanding of human NER, we modified the model of Kesseler et al. to include mechanistic kinetic proofreading. The random order cooperative binding of the first three repair factors was retained as in the original model, and kinetic proofreading was introduced as illustrated in the schematic to the right. The modified model differs in a number of important ways, as can be seen in the schematic. First, PIC3 is included as a stable species rather than a surrogate for excised DNA, and a final irreversible excision step with rate constant k_cut was added. This was necessary because rather than couple the proofreading to the reactions, we coupled it to the complexes, which more accurately reflects the time delay nature of kinetic proofreading. Each of the three PICs can go forward to the next PIC or can dissociate fully to start the process over again. The parameters α, β, and γ represent the k_off's for PIC dissociation, and are dependent upon the substrate (damaged or undamaged). Our model thus introduces three new species (PIC3 for repair factors assembled at a lesion, away from a lesion, and on an undamaged strand) and seven new parameters (dissociation rates for PIC1, PIC2, and PIC3 on damaged and undamaged DNA, and k_cut). Parameter values were initially determined empirically to recapitulate the results of in vitro assays by Kesseler et al., but because they are biologically based, they will be determined experimentally in Specific Aim 2. Parameters and their empirical values are shown below.
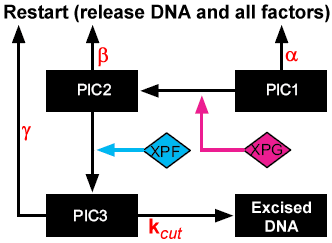{kind=link}
| Paramter | Description | Value |
|---|---|---|
| α | Dissociation of PIC1 from damaged DNA | 9 x 10^-8 (s^-1) |
| β | Dissociation of PIC2 from damaged DNA | 8 x 10^-8 (s^-1) |
| γ | Dissociation of PIC3 from damaged DNA | 8 x 10^-4 (s^-1) |
| -α | Dissociation of PIC1 from undamaged DNA | 1 (s^-1) |
| -β | Dissociation of PIC2 from undamaged DNA | 3 (s^-1) |
| -γ | Dissociation of PIC3 from undamaged DNA | 0.11 (s^-1) |
| k_cut | Excision from PIC3 | 0.0072 (M^-1 s^-1) |
D.2 Obtain biologically-based model parameters through experimental manipulation
The major improvement of our model over existing ones is that our parameters are biologically meaningful and therefore testable. The standard in vitro assay for NER relies on reconstituting the system in cell-free extract (CFE) using six recombinantly expressed NER factors (RPA, XPA, XPC-TFIIH, XPG, and XPF) [9,10]. Note that while XPC and TFIIH are separate proteins, their affinity is such that they are almost never found not in complex [3]. An artificial substrate is provided in the form of a synthetic double-stranded oligonucleotide, typically ~150-bp in length [11]. Together these three components are sufficient to reconstitute NER. The relative simplicity of this system combined with the well understood, manipulable genetics of the protein components (the system has been effectively reconstituted with proteins recombinantly expressed in in S. cerevisiea [12]) makes it amenable to manipulation, as we propose below. The system also permits the use of a standard NER assay, as described previously [3]. Briefly, a ~150-bp oligonucleotide of radiolabeled DNA containing an adduct known to induce NER (obtained commercially or synthesized in-house) is incubated with CFE and repair proteins for a set amount of time, then DNA is isolated and analyzed by polyacrylamide gel electrophoresis and autoradiogram to visualize the 24-32 nt excision product [7]. This assay allows us to quantify both the extent and the efficiency of repair.
Our kinetic assays are based on a recently perfected system that uses real time fluorescent microscopy with single molecule resolution to track the assembly and disassembly of protein complexes on DNA [13]. The system uses short oligonucleotides bound to a surface in a functionalized microfluidic channel (by biotin-streptavidin interaction). Buffer is flowed through the channel at a rate such that the resulting force causes the DNA to stretch to its normal length. A fluorescently tagged version of the protein of interest is then introduced to the buffer, and the system is recorded in real time by single-molecule resolution fluorescent microscopy. This permits direct visualization of the assembly and disassembly of the DNA-protein complexes. Tracking the gain and loss of fluorescent signal allows quantitative kinetic data to be extracted [34]. This system was previously used to determine the k_off for Rad51, a DNA binding protein involved in the homologous recombination (HR) repair pathway [35]. Given the similarities in complexity and protein function between NER and HR, we are confident that this system will permit the accurate determination of the model parameters α, β, and γ.
We will first create fluorescently labeled versions of the repair proteins XPC, XPG, and XPF. Entry of each of these proteins is associated with certain transition in the repair pathway. Specifically, XPC binds to form PIC1, XPG binds to form PIC2, and XPF binds to form PIC3. Both biological fluorescence (protein fusions with cyan fluorescent protein) and chemical fluorescence (Alexa Fluor dyes chemically conjugated) will be synthesized and assayed for function against the corresponding WT protein using the standard NER assay. The tag showing the least effect on protein function will be used for all analyses.
We will then obtain biotinylated oligionucleotides containing a single fluorescent adduct; the exact design of this adduct will be optimized experimentally, but one possibility is a Alexa Fluor dye conjugated to an exocyclic amine in a purine, similar in structure to the benzopyrene adduct that is known to induce NER. Using a fluorescent adduct permits visual identification and isolated tracking of only repair proteins that bind at the site of damage. Again, all constructs will be subjected to the standard NER assay with both WT and fluorescent proteins as well as both normal and fluorescent DNA adducts. This level of control experiments is necessary to prove that the modification to the system do not perturb the kinetics, which would invalidate our analyses.
The complete design of the assay is as follows. Commercially obtain a custom microfluidic chamber functionalized with streptavidine, and introduce biotinylated oligonucleotides. Flow a buffer containing CFE and all proteins except the appropriate fluorescent one through the channel. Simultaneously introduce the fluorescent protein and a non-hydrolyzable ATP analogue (β,γ-methylene adenosine 5'-triphosphate, AMP-PCP). Under normal conditions, PIC dissociation can occur because of progressing to the subsequent complex (ATP-dependent) or dissociating fully (kinetic proofreading, ATP-independent). Introduction of AMP-PCP competitively abrogates the transition between PICs and allows for isolation of the ATPindependent dissociation mechanism. Track the reaction progress by real time fluorescent microscopy, following a single oligonucleotide where the repair protein has bound to the site of damage. The fluorescent intensity can then be plotted as a function of time and fit to a single exponential decay function, allowing k_off to be extracted as previously described [35]. This assay will be performed a total of six times: three times (once for each repair protein) with adducted DNA and three with undamaged DNA, yielding the first six parameters α, β, γ, -α, -β, and -γ.
To determine the final parameter, the rate constant for excision from PIC3 (designated k_cut), a similar assay using commercially available molecular beacon probes will be utilized. A molecular beacon is a short (~25-nt), single-stranded oligonucleotide that is complimentary to a sequence of interest with a fluorophore and a quencher attached at opposite ends [14]. In solutoin, the probe forms a hairpin loop, quenching the fluorescence. When the probe encounters a complementary sequence of single stranded DNA/RNA, it hybridizes to the target and fluoresces [18]. The change from hairpin to open form is mediated solely by the increased stability of the hybridized form, and does not require heating of the reaction mixture, which greatly decreases the potential for cross-reactivity. In addition, the difference in energy between the hairpin and the hybrid can be "tuned" finely enough that the beacon can effectively discriminate between a perfect target sequence and one harboring a single mismatch [16]. Molecular beacons are therefore ideally suited for detecting the appearance of known singlestranded DNA sequences in real time [15].
The complete assay is as follows. Commercially obtain a molecular beacon specific for 25-nt sequence containing the damage product in the synthetic oligonucleotide. Assemble all NER proteins except XPF in CFE. After a sufficient delay to allow PIC2 complexes to form, simultaneously add XPF and the molecular beacon, inducing excision. Monitor the reaction by real-time fluorescence microscopy, and the time course (appearance of signal) will correspond to the transition from PIC3 to excised DNA, allowing k_cut to be extracted. To ensure that the presence of the molecular beacon does not alter the repair activity, a control experiment will be performed to compare repair (as described above) in a WT system and in WT + molecular beacon.
D.3 Validate model hypotheses in an in vitro cell-free model
The results of our preliminary modeling have generated multiple testable hypotheses regarding the stabilities and affinities of various complexes and proteins in the NER pathway. Specifically, our model indicates that the PIC3 complex bound to damaged DNA is significantly more stable than either PIC1 or PIC2, whereas the PIC1 complex bound to undamaged DNA is significantly more stable than either PIC2 or PIC3. Because each PIC is characterized by one unique protein (XPC for PIC1, XPG for PIC2, and XPF for PIC3), it is likely that the relative affinity of each of these proteins for DNA is the principle determinant of the corresponding PIC's stability. Protein affinity for DNA can be accurately measured with surface plasmon resonance (SPR) [19].
We will first determine the affinity (K_d) of XPC, XPG, and XPF for both damaged and undamaged DNA. We will use short, synthetic oligonucleotides similar to those described in D.2, conjugated to a gold surface as previously described [20]. Because it is possible for the various NER proteins to bind to undamaged DNA, the oligos used to determine affinity for damaged DNA will contain multiple identical adducts spaced at regular intervals (the exact distance between them will need to be optimized). This will decrease the probability that the proteins will bind away from the site of damage. Purified, recombinantly expressed NER proteins will be flowed over the gold films (one protein per assay) and SPR will be applied, allowing us to extract the K_d for each protein by global parameter fitting [21]. Based on our model, we expect XPC to have the highest affinity for undamaged DNA and XPF to have the highest affinity for damaged DNA. If this is not the case, it would provide evidence for an alternative hypothesis in which the stability of the PIC is mediated by the interaction between the unique protein and the others in the complex, perhaps through a conformational change. Such a hypothesis could easily be tested by experiments similar to those described below for our initial hypothesis.
To determine if the affinity of the repair proteins for DNA significantly affects the progress of NER, we will create mutant proteins with decreased affinity for DNA. The structures and DNA binding motifs for all three proteins are well-characterized at the genetic level [22-24]. We will utilize PCR-mediated, site-directed mutagenesis to introduce a variety of mutations into the DNA binding motif of the proteins identified as having the highest affinity for damaged and undamaged DNA [25]. These mutated proteins will be recombinantly expressed and their DNA affinity determined by SPR. Mutants with reduced affinity will then be combined with other WT proteins in the standard NER assay to determine if they inhibit repair (using all WT proteins as a control). These assays will be conducted with both damaged and undamaged DNA (separate assays) to test both hypotheses: that they display defective NER and that they display increased excision of undamaged DNA (compared to WT control in both cases).
D.4 Validate in vitro predictions in an in vivo whole-cell system
The final test of the functional studies in D.3 will be to confirm the predicted phenotype in a whole-cell system. Specifically, our model predicts that cells harboring the mutant forms of XPC and XPF described above should display increased sensitivity to chemical- and UV-induce DNA damage. This hypothesis will be tested using a variety of established techniques.
Human cell lines deficient in each NER repair protein are commercially available, as are a number of mammalian vectors for introducing recombinant mutated protein into these cells [26]. Cells lacking each of the proteins mutated in D.2 will be obtained and transfected with our corresponding low affinity mutant protein. Control cells will be transfected with corresponding WT protein. Cells will be exposed to UV radiation or a chemical known to cause NER-specific adducts (such as benzopyrene-diol-epoxide), according to established dosing protocols [27]. Cell survival will be assayed as the phenotypic indicator of sensitivity, and dose-response curves will be constructed for both WT and mutant cells. Again, based on our hypothesis we predict that both mutants will display increased sensitivity to UV and chemical damage, as compared to WT cells. Analysis of the precise mechanism of death in sensitive cells (accumulation of damage, excision of undamaged DNA, etc.) will constitute the first step in determining potential therapeutic targets.
References
- E. Evans, J.G. Moggs, J.R. Hwang, J.-M. Egly, & R.D. Wood. The EMBO Journal 1997, 16: 6559-6573
- K.J. Kesseler, W.K. Kaufmann, J.T. Reardon, T.C. Elston, & A. Sancar. Journal of Theoretical Biology 2007, 249: 361-375
- J.J. Hopfield. PNAS 1974, 71: 4135-4139
- J.T. Reardon & A. Sancar. PNAS 2006, 103(11): 4056-4061
- J.T. Reardon & A. Sancar. Molecular & Cellular Biology 2002, 22(16): 5938-5945
- D.B. Zamble, D. Mu, J.T. Reardon, A. Sancar, & S.J. Lippard. Biochemistry 1996, 35: 10004-10013
- W. Yang (ed). DNA Repair & Replication. Academic Press 2004.
- S.-Ullah, I. Husain, W. Carlton, & A. Sancar. Nucleic Acids Research 1989, 17(12): 4471-4484
- J.T. Reardon & A. Sancar. Genes and Development 2003, 17: 2539-2551
- S.J. Araujo, F. Tirode, F. Coin, H. Pospiech, J.E. Syvaoja, M. Stucki, U. Hubscher, J.-M. Egly, & R.D. Wood. Genes and Development 2000, 14: 349-359
- S. Prakash & L. Prakash. Mutation Research 2000, 451: 13-24
- J. Hilarioab, I. Amitaniab, R.J. Baskinb, & S.C. Kowalczykowski. PNAS 2009, 106(2): 361-368
- W. Tan, K. Wang, & T.J. Drake. Current Opinions in Chemical Biology 2004, 8(5): 547-553
- S.A.E. Marras, F.R. Kramer, & S. Tyagi. Genetic Analysis: Biomolecular Engineering 1999, 14(5-6): 151-156
- T. Drake & W. Tan. Applied Spectroscopy 2004, 58: 269-280
- E. Evans, J. Fellows, A. Coffer, & R.D. Wood. The EMBO Journal 1997, 16: 625-638
- S. Tyagi & FR Kramer. Nature Biotechnology 1996, 14: 303-308
- J. Homola. Analytical and Bioanalytical Chemistry 2003, 377(3): 528-539
- J.M. Brockman, A.G. Frutos, & R.M. Corn. Journal of the American Chemical Society 1999, 121: 8044-8051
- D.G. Myszka. Current Opinions in Biotechnology 1997, 8(1): 50-57
- L. Li, C. Peterson, & R. Legerski. Nucleic Acids Research 1996, 24(6): 1026-1028
- S. Emmert, T.D. Schneider, S.G. Khan, & K.H. Kraemer. Nucleic Acids Research 2001, 29(7): 1443-1452
- J.H. Enzlin & O.D. Scharer. The EMBO Journal 2002, 21: 2045-2053
- R. Prasad, W.A. Beard, J.Y. Chyan, M.W. Maciejewski, G.P. Mullen, & S.H. Wilson. Journal of Biological Chemistry 1998, 273(18): 11121-11126
- L. Li, C.A. Peterson, X. Lu, & R.J. Legerski. Molecular and Cellular Biology 1995, 15(4): 1993-1998
- M. Wakasug, A. Kawashima, H. Morioka, S. Linn, A. Sancar, T. Mori, O. Nikaido, & T. Matsunaga. Journal of Biological Chemistry 2002, 277(3): 1637-1640
- F.R. de Gruijl, H.J. van Kranenc, & L.H.F. Mullenders. Journal of Photochemistry and Photobiology B: Biology 2001, 63(1-3): 19-27
- A.L. Jackson & L.A. Loeb. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis 2001, 477(1-2): 7-21
- M.T. Hess, D. Gunz, N. Luneva, N.E. Geacintov, & H. Naegeli. Molecular and Cellular Biology 1997, 17(12): 7069-7076
- J.T. Reardon, T. Bessho, H.C. Kung, P.H. Bolton, & A. Sancar. PNAS 1997, 94(17): 9463-9468
- W.L. de Laat, N.G.J. Jaspers, & J.H.J. Hoeijmakers. Genes and Development 1999, 13: 768-785
- J.T. Reardon & A. Sancar. Cell Cycle 2004, 3(2): 141-144
- C. Joo, S.A. McKinney, M. Nakamura, I. Rasnik, S. Myong, & T. Ha. Cell 2006, 126(3): 515-527
- M. Modesti, D. Ristic, T. van der Heijden, C. Dekker, J. van Mameren, E.J.G. Peterman, G.J.L. Wuite, R. Kanaar, & C. Wyman. Structure 2007, 15(5): 599-609